|
The Retina is a Part of the Brain
Visual perception begins when light from our surroundings enters our eyes and
passes through the transparent cornea and lens. The curvatures of the cornea and
lens bend the rays of light, like the lens of a camera, and bring them into focus
upon a thin, transparent layer of nerve tissue, called the retina, that lines
the inner surface of the eye.
The retina is actually part of the central nervous system. It is connected to
the brain by a cable called the optic nerve that is made up of approximately one
million nerve fibers. The retina contains a variety of specialized cell types
called neurons, and supporting cells called glia, which are interconnected in
various ways at contact points known as synapses and junctions. Six major cell
classes, and more than 50 distinct neuronal cell configurations, have been identified
in the retina to date. Photoreceptor cells make up the bulk of the neurons in
the retina. Each retina contains approximately 105 million photoreceptor cells
whose job it is to capture units of light energy called photons and to register
those events as electrical signals in the "language" of the central
nervous system. The electrical signals generated by the photoreceptors are then
relayed to intermediary layers of neurons in the retina that process and organize
this information before it is transmitted along the optic nerve fibers to the
brain.
What is Macular Degeneration?
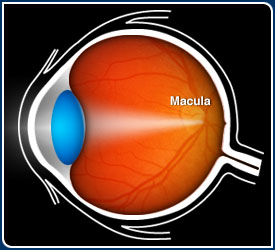
Macular degeneration is a blinding disease caused by the death of the photoreceptor
cells in that part of the retina known as the macula. The macula is a circular
area, approximately 3 mm or about 1/10 inch in diameter, that is located next
to the optic nerve. In the age-related form of macular degeneration (ARMD), photoreceptor
cells within the macula die off slowly, thus accounting for the progressive loss
of vision that usually begins after the fifth or sixth decade of life.
The two main classes of photoreceptors, rods and cones, are named from their cylindrical
or conical shapes respectively. In general, the rod system functions primarily
at low light levels (i.e. at night), whereas the cone system is designed to detect
color and fine detail under daylight conditions. The majority of photoreceptor
cells in the macula are cones, whereas most photoreceptors in outlying regions
of the retina are rods. As one moves toward the center of the macula, the percentage
of cones rises sharply, and reaches 100% in the center of a specialized region
called the fovea. It is the degeneration of photoreceptor cells, primarily in
the macula, that accounts for the loss of central vision and fine detail vision
that defines ARMD.
The precise reason(s) why the photoreceptors die in ARMD has puzzled vision researchers
for many decades, and numerous theories have been advanced to account for this
phenomenon. In addition to a general decline in the functional capacity of tissues
and organs that is an intrinsic part of aging, genetic defects and environmental
factors including nutritional deficiencies, excessive light exposure, and inadequate
blood supply have all been invoked to explain their death. Thus far, however,
there is no definitive and compelling evidence for any single theory.
Gene Discovery Research on Macular Degeneration
There is a scientific consensus that ARMD has a number of associated
risk factors, one of which is a family history of the disease. This means
that ARMD is similar to other diseases of aging, such as Alzheimer's disease and
atherosclerosis, where a history of disease in one or more family members puts
an individual at increased risk of developing it at some point in his/her lifetime.
Furthermore, studies of identical and fraternal twins have shown convincingly
that the development of ARMD in one sibling puts the other sibling at very high
risk. These facts, together with the advent of modern molecular genetics, has
triggered an intensive search for the gene(s) that may increase one's susceptibility
to ARMD later in life.
The study of families with a high incidence of other macular diseases that usually
arise much earlier in life, well before the age of 50, has led to the identification
of several genes that cause certain rare forms of inherited macular degeneration.
Click Here for
a comprehensive list of genes linked to retinal degenerative diseases. These diseases
include pattern dystrophy, Best disease, Stargardt disease, Stargardt-like dominant
macular dystrophy, Sorsby fundus dystrophy, and Malattia Leventinese (also known
as Doyne's honeycomb retinal dystrophy). Although these other macula disorders
are not the same as ARMD, the similarities are sufficient to warrant additional
research to determine whether one or more of the same genes could also be involved.
Recently, genetic linkage studies of individuals with and without ARMD have identified
a candidate gene that could account for a small percentage of the more prevalent,
age-related form of macular degeneration. This has led many to question whether
ARMD is really a single entity or, like other retinal diseases such as Retinitis
Pigmentosa, a family of related disorders with a genetic component that is characterized
by a similar pattern of visual symptoms. The intensive search for ARMD-related
genes continues and, with the completion of the Human Genome Project, should accelerate
into the foreseeable future. It can be anticipated with some confidence that additional
genes will be identified within the next few years that will be linked to the
development of different forms of ARMD.
Cell and Molecular Biological Research on Macular Degeneration
The search for ARMD genes is augmented by research on the cell biology of this
disease. The ultimate goal of this research is to identify the molecular mechanisms
and cellular events that are responsible for photoreceptor cell death. This approach
utilizes a variety of modern cell and molecular biological techniques, biochemical
methods, and microscopic instrumentation. With the possible exception of some
aged, non-human primates, animals do not develop ARMD. Therefore blood, ocular
tissue specimens, and cells obtained from human donors provide the only available
resource for such studies.
What Cells and Tissues are Involved?
Ophthalmologists have known for many years that abnormalities in the appearance
of the retina can be used to diagnose and, in some cases, predict those who will
go on to develop the more severe, vision-threatening forms of ARMD. Post-mortem
examination of the eyes of individuals who show such changes using the light and
electron microscopes have identified the anatomical location where those abnormalities
occur. The affected region lies at the boundary between the photoreceptors cells
of the retina and the choroid, a layer of connective tissue that contains the
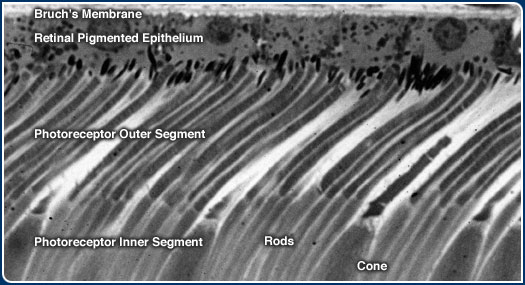
blood vessels which supply the photoreceptors. This region includes a single layer
of pigmented cells, known as the retinal pigmented epithelium (RPE) that are anchored
to a multilayered meshwork of fibrous proteins known as Bruch's membrane. The
earliest recognizable signs of ARMD occur at the interface between Bruch's membrane
and the RPE.
Photoreceptors are Dependent upon Supporting Cells for Survival
The photoreceptor cells are among the most metabolically active cells in the body.
As such, they are critically dependent upon a network of blood vessels, one in
the retina and another in the choroid, for the delivery of oxygen and other essential
nutrients, such as vitamin A, that circulate continuously in the blood. The RPE
occupies a strategic location between the blood vessels in the choroid and the
photoreceptors. Thus, these cells are in a unique position to mediate the transport
and clearance of blood borne nutrients and other metabolites to, and from, the
photoreceptors.
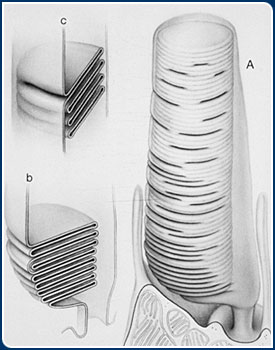
Several decades ago, it was discovered that the RPE cells are responsible for
the removal and digestion of molecular components that are shed from one end of
both rod and cone photoreceptor cells on a daily basis. This portion of the photoreceptor
is known as the outer segment and it consists of a stack of membranes, similar
to a roll of coins. The outer segment houses the molecular machinery responsible
for capturing photons of light that impinge upon the retina. The outer segment
membranes contain a light-absorbing protein called opsin which is tethered to
a molecule of vitamin A. When photons strike the opsin-vitamin A complex, it undergos
a change in shape which, in turn, triggers a cascade of biochemical reactions
all of which occur in a small fraction of a second. This fundamental process,
called visual transduction, initiates the biochemical reactions that ultimately
lead to the perception of a visual image. The biochemical cascade leads to the
generation of an electrical signal that is transmitted to the opposite end of
the photoreceptor cell, and from there to neurons in the inner retina for processing
, before it is relayed to processing centers in the brain via the optic nerve.
Photoreceptor outer segment membranes are manufactured continuously at the base
of the stack and, periodically, shed from their tips. In a process known as phagocytosis,
small packets of the shed membranes are then enveloped by tentacles of the RPE
cells, and incorporated into the cells' interior where they are digested by enzymes.
This mechanism of photoreceptor outer segment membrane turnover is also an indispensable
part of the process required for normal vision.(Quick
Time Movie of how photoreceptor outer segment renewal works). In the absence
of a viable and healthy RPE, the shed photoreceptor membranes cannot be removed
and digested promptly, membranous debris builds up, and the photoreceptor cells'
supply of vitamin A and other nutrients becomes depleted. If such conditions persist,
the photoreceptors die.
What is the Primary Defect in ARMD?
Because the photoreceptors are critically dependent upon the RPE cells for their
own survival, it is not clear whether the primary defect(s) in ARMD lies in the
photoreceptor cells, in the RPE cells or, perhaps, in some combination of both.
It is known, however, that genetic alterations (i.e. mutations) in rod photoreceptor-specific
genes, including the rod opsin gene, are responsible for a separate family of
blinding disorders characterized primarily by the loss of rods and peripheral
vision. By analogy, mutations in one or more cone opsin genes are obvious candidates
as ARMD genes, although there is no published evidence of their involvement thus
far. At least one other photoreceptor-related gene, ABCA4, is under investigation
as a candidate ARMD gene at the present time. Mutations in the ABCA4 gene cause
Stargardt's disease, a rare inherited form of macular degeneration that occurs
in one person out of every 10,000.
Many vision researchers believe that the visual deficits that account for a significant
proportion of ARMD are not caused by photoreceptor gene alternations. According
to this theory, photoreceptor cell death is a secondary consequence of a primary
defect in the RPE cells, which could be attributable to any number of genetic
and/or environmental factors. As such, it is appropriate that the study of the
RPE in both health and disease has been, and continues to be, the object of intensive
research.
Deposits called Drusen are associated with ARMD
By the age of 60 in most individuals, prominent changes take place in the RPE
cells, and along Bruch's membrane, the platform on which they are anchored.
The cells become increasingly laden with pigment granules called lipofuscin that
contain concentrated by-products of cellular metabolism which are potentially
toxic, highly resistant to further degradation, and therefore difficult to eliminate.
Bruch's membrane, which is rich in fibrous proteins such as collagen and elastin,
thickens significantly and several types of deposits start to accumulate, each
of which is associated with a specific layer of Bruch's membrane.
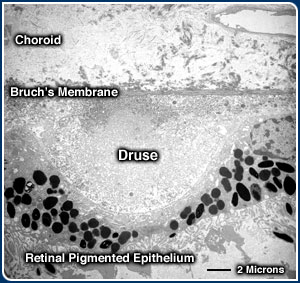
The most prominent of these deposits are called drusen: small, balloon-like expansions
of the innermost layer of Bruch's membrane that displace the overlying RPE and
photoreceptors cells.
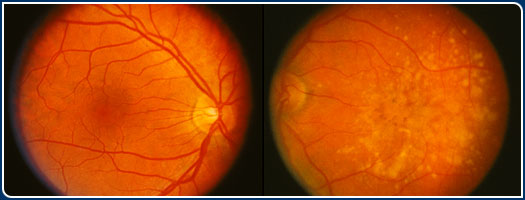
When an ophthalmoscope is used to visualize the macula at rear of the eye, drusen
appear as small yellow spots that range in size from 30 to over 100 micrometers
in diameter, or between 1/1000 to 4/1000 of an inch. Electron microscopic examination
of tissue sections (see below) reveals many more drusen within and outside of
the macula that are up to 10 times smaller.
Drusen are regarded by many ophthalmologists as the hallmark clinical sign of
early ARMD. The appearance of numerous drusen and/or large drusen, especially
in the macular region, is a significant risk factor for the subsequent development
of the most prevalent type of ARMD, the atrophic or "dry" form, that
accounts for approximately 85% of clinically diagnosed ARMD. Identifying the origin
and molecular composition of these drusen deposits, therefore, has remained an
important but elusive objective for many decades.
|